Responsabili: Dott.ssa M.G. D’Angelo e Dott.ssa F. Magri
Lavoro collaborativo tra IRCCS E. Medea di Bosisio Parini e Fondazione Policlinico di Milano- Clinica
Negli ultimi anni la risonanza magnetica (RM) muscolare ha giocato un ruolo sempre più determinante sia in ambito diagnostico sia in ambito di valutazione di efficacia terapeutica nelle patologie neuromuscolari e nelle patologie neuromotorie.
Nelle malattie muscolari lo studio tramite RM muscolare può, tramite l’identificazione di pattern specifici di interessamento muscolare, guidare verso una più immediata diagnosi differenziale in ambito genetico e biochimico. Lo studio specifico inoltre della muscolatura respiratoria e la sua correlazione con parametri funzionali può fornire strumenti utili a scopo predittivo o di valutazione di efficacia di alcuni trattamenti (R-Y Carlier, 2012; Hollingworth KG, 2012).
In particolare la RM muscolare è stata oggetto di numerosi studi, soprattutto negli ultimi anni.
Recenti lavori, principalmente effettuati con RM a 1.5T (Hollingsworth KG et al 2012, Astrea G 2009, Astrea G 2012), hanno consentito di definire dei pattern specifici di interessamento muscolare nelle diverse forme di distrofia muscolare suggerendo la possibile applicazione di questo strumento per guidare la diagnosi differenziale. Al momento attuale la maggior parte dei lavori ha indagato l’interessamento degli arti inferiori, anche se alcuni studi di RM whole body sono stati pubblicati (Kesper K et al., 2008).
Inoltre pochi sono gli studi longitudinali finora condotti (Willis TA et al. 2013) L’acquisizione di dati longitudinali potrebbe invece essere fondamentale al fine di valutare l’efficacia di questo strumento per tracciare la storia naturale di malattia e come misura di outcome nella valutazione di efficacia in potenziali futuri studi farmacologici.
- Lo Studio
- Background
- Obiettivi
- Campione
- Attività Consenso
- Attività Caratterizzazione
- Attività Imaging
- Dati Preliminari
Lo studio si propone di selezionare 20 pazienti affetti da Distrofia Muscolare dei Cingoli (suddivisi per tipologia sulla base del gene coinvolto), 5 pazienti affetti da Miopatia congenita e 5 pazienti affetti da Distrofia Congenita. Il nostro proposito è quello di studiare i pazienti affetti da distrofia muscolare ad un tempo 0 ed in tempi successivi a distanza di 12 e 24 mesi.
Verranno effettuate acquisizioni a 3 T della muscolatura del cingolo scapolare e della muscolatura prossimale degli arti superiori, utilizzando sequenze pesate in T1, T2, inversion recovery e 3 point Dixon (Hollingsworth KG et al 2012, Astrea G 2009, Astrea G 2012). Il grado di interessamento muscolare verrà valutato con criteri semiquantitativi utilizzando scale specifiche (Mercuri E. et al, 2002). In un sottogruppo di pazienti verranno inoltre utilizzate sequenze specifiche in grado di valutare la perfusione muscolare, al fine di definire la presenza di una compromissione di perfusione e di compromissione metabolica in alcuni tipi di patologie muscolari studiate.
In un sottogruppo limitato di pazienti condurremo inoltre uno studio esplorativo al fine di andare a valutare la sensibilità e specificità dello studio di Risonanza Magnetica della muscolatura intercostale, addominale e diaframmatica e l’eventuale correlazione con la funzionalità respiratoria.
Nel gruppo di soggetti affetti invece da patologie neuromotorie quali le paraparesi spastiche ereditarie e le paralisi cerebrali infantili, ci prefiggiamo di definire la morfologia di un muscolo eziopatogenicamente integro da un punto di vista strutturale ma sottoposto a stress di origine “centrale” e di definire il ruolo su di esso di trattamenti miorilassanti locali come la tossina botulinica ed eventuale identificare agenti modulatori di diversa efficacia.
Studieremo tramite RM muscolare (ipotizzando studio di muscolo gastrocnemio e quadricipite femorale) 5 soggetti affetti da paraparesi spastica ereditaria (HSP) e 5 soggetti con PCI mai trattati con tossina botulinica e rispettivamente 5 soggetti HSP e 5 soggetti PCI già precedentemente trattati, pre e post trattamento con tossina botulinica.
Le malattie neuromuscolari sono un gruppo clinicamente e geneticamente eterogeneo di patologie caratterizzate da progressiva ed ingravescente debolezza muscolare spesso associata ad un interessamento cardiaco, respiratorio e cognitivo. Attualmente non vi sono terapie efficaci e più del 50% di queste forme è ancora priva di diagnosi molecolare, a causa della complessità del processo diagnostico. Una corretta definizione delle basi molecolari è tuttavia un prerequisito essenziale per il management clinico, per approfondire lo studio della patogenesi e per formulare ipotesi terapeutiche.
Recentemente lo sviluppo di nuove strategie diagnostiche quali l’analisi genetica tramite Next-generation Sequencing o l’applicazione della Risonanza Magnetica muscolare hanno affinato la capacità diagnostica per questo gruppo di patologie, portando ad interessanti risultati e a una migliore comprensione della patogenesi, sebbene entrambe le tecniche citate non vengano ancora applicate di routine.
Obiettivo Principale
Obiettivo principale dello studio è quello di migliorare il processo diagnostico di alcune delle principali forme di malattie neuromuscolari e di approfondirne i meccanismi patogenetici. Lo studio tramite RM muscolare è principalmente volto ad identificare pattern specifici di interessamento muscolare che potranno essere utilizzati per guidare la diagnosi differenziale e l’evoluzione della malattia. Lo studio tramite RM della muscolatura respiratoria e la sua correlazione con parametri funzionali potrà fornire strumenti utili a scopo predittivo o per valutare tale funzione delle differenti patologie nelle diverse fasi di progressione.
Obiettivo Secondario
Valutazione longitudinale di un gruppo di pazienti affetti da distrofia dei cingoli definiti dal punto di vista genetico. Le valutazioni della funzionalità motoria con scale riconosciute a livello internazionale (Motor Function Measure scale) e 6 minute walk test (in pazienti ancora deambulanti) e le valutazioni della forza muscolare tramite scala MRC verranno comparate ai dati di imaging ottenuti. Verrà valutata l’utilità dell’imaging muscolare nel follow-up delle diverse forme e come potenziale out come clinico per futuri trial terapeutici, in possibile sostituzione della biopsia muscolare.
Lo studio in oggetto è uno studio esplorativo, volto a definire il ruolo e la sensibilità della RM nel valutare il grado di interessamento della muscolatura scheletrica e respiratoria in soggetti con sofferenza muscolare primitiva e secondaria. Fino ad oggi gli studi effettuati e pubblicati in letteratura si basano su piccolo campioni di pazienti.
Verrà studiato un campione complessivo di 50 soggetti, a sua volta suddiviso in diversi gruppi a seconda della patologia considerata. Nel complesso per ogni tipo di patologia verranno valutati almeno 5 pazienti con diverso grado di compromissione muscolare. Reputiamo che tale numero di pazienti sia sufficiente per consentire uno studio comparativo e definire eventuali differenze tra i sottogruppi studiati. Sulla base dei risultati ottenuti potrà essere deciso di ampliare ulteriormente la numerosità del campione.
Acquisizione del consenso informato
In questo primo anno abbiamo proceduto, dopo raccolta di Consenso Informato, con la caratterizzazione clinica dei pazienti tramite:
• raccolta di dati anamnestici e dati relativi alla presenza di eventuale familiarità e dei dati relativi alla diagnosi comprensivi di dati di analisi genetica, biochimica ed istologica muscolare.
• dettagliata valutazione neurologica effettuata tramite misure di forza (scala MRC) ed esecuzione di scale funzionali quali la Motor Function Measure (MFM) scale e il 6 minute walk test (effettuato in paziente ancora deambulanti) e,laddove possibile, anche la funzionalità degli arti superiori attraverso la scala Upper Limb.
Tutti i pazienti inoltre sono stati sottoposti anche a :
• valutazione della funzionalità respiratoria (tramite spirometria, registrazione di saturimetria notturna, e pletismografia optoelettronica) e della funzionalità cardiaca (tramite ecocardiogramma, elettrocardiogramma -ECG e, laddove necessario, registrazione di ECG-holter).
Caratterizzazione clinica dei pazienti
Tutti i dati sono inseriti in un sistema DATABASE informatizzato (la cui creazione è avvenuta grazie ad un progetto di Ricerca Corrente degli scorsi anni), con accesso limitato da password; il “database neuromuscolare” è un’applicazione web disegnata per raccogliere e riportare I dati raccolti di pazienti affetti da patologie neuromuscolari .
Il software è stato sviluppato con linguaggio PHP, utilizzando il “RadPHP” framework, mentre i dati sono archiviati nel database relazionale PostgreSQL; sia l’applicazione PHP sia il motore del database sono ospitati nel sever Linux. L’impiego di tecnologie standard Html, Css e Javascript per creare l’interfaccia dell’utente rende l’applicazione accessibile a tutti gli utenti con un sistema di navigazione Web, senza aver bisogno di un’installazione locale di altre applicazioni.
Grazie a questo approccio via Web, l’accesso al sistema non è limitata alle stazioni informatiche della nostra Struttura ma può essere gestita su PC esterni all’Istituto, con l’obiettivo di facilitare la raccolta dati dello stesso paziente da strutture sanitarie diverse.
E’ presente sistema di tracciatura degli accessi utente, con gestione profili; ogni utente è associato ad uno specifico profilo che determina l’accessibilità delle varie schede in sola lettura o in scrittura sulla base delle competenze professionali e dei protocolli di studio per i quali il Database viene utilizzato.
Sono infine stati disaccoppiati i dati anagrafici dai dati clinici per consentire il trattamento anonimo dei dati personali dei pazienti per elaborazioni ed analisi statistiche.
I dati possono essere aggregati e filtrati con l’obiettivo di ottenere informazioni su un singolo paziente (ad esempio con l’obiettivo di valutare l’evoluzione di alcuni parametri nel singolo soggetto) oppure su un gruppo di pazienti. I risultati possono essere agevolmente salvati ed importati su un file Excel (o altri fogli elettronici) o su sistemi di analisi statistica come SPSS, dove l’utente può utilizzare le informazioni per creazione di grafici o analisi statistiche.
Di seguito riportiamo alcuni esempi: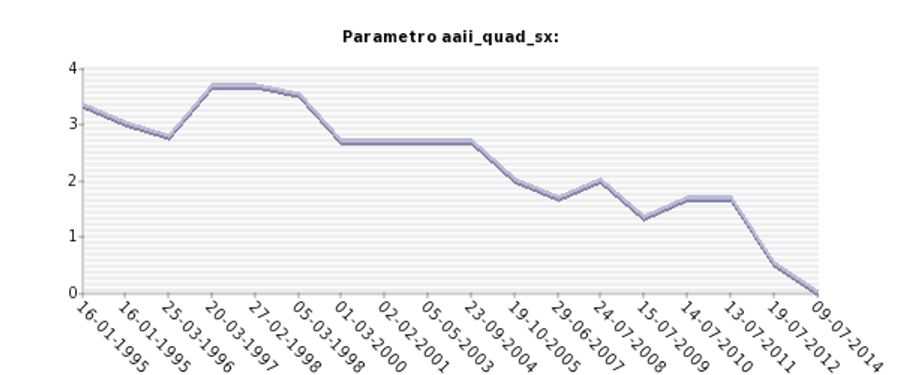
Grafico ottenuto dal database: Analisi longitudinale della stenia del muscolo quadricipite femorale sinistro , misurata tramite scala MRC (0-5) in un paziente affetto da distrofia muscolare dei cingoli da deficit di calpaina 3
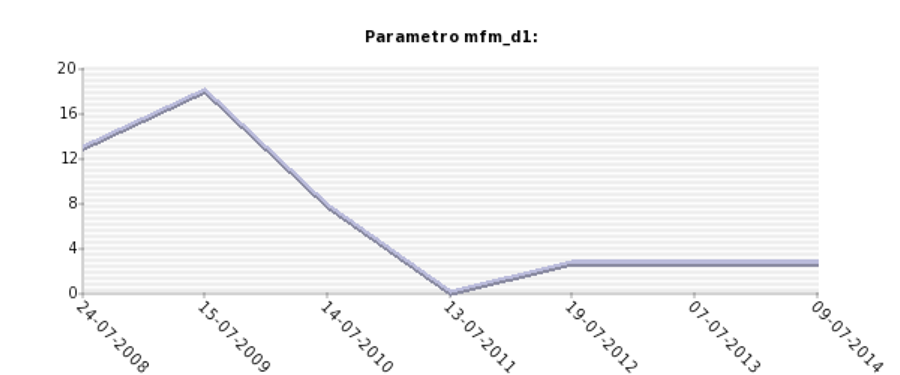
Grafico ottenuto dal database: Analisi longitudinale della valutazione funzionale motoria tramite scala Motor Function Measure, subscala D1 (stazione eretta/trasferimenti), misurata tramite scala MRC (0-5) in un paziente affetto da distrofia muscolare dei cingoli da deficit di calpaina 3
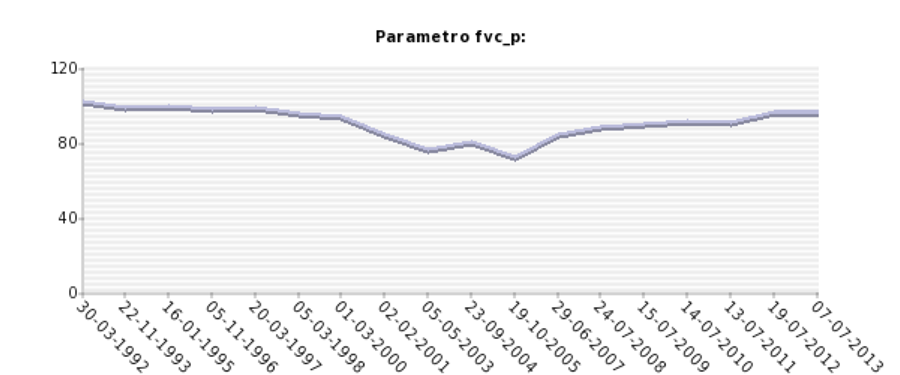
Grafico ottenuto dal database: Analisi longitudinale della valutazione funzionale motoria tramite spirometria (parametro FVC) in un paziente affetto da distrofia muscolare dei cingoli da deficit di calpaina 3
Grazie ai dati clinici raccolti, abbiamo iniziato un’analisi volta ad identificare sia in ogni singolo soggetto sia nel gruppo di appartenenza del singolo (ad esempio gruppo delle distrofie muscolare dei cingoli da deficit di calpaina 3-LGMD2A).
Andando a valutare i dati ottenuti alle scale muscolari (MRC), abbiamo identificato il muscolo/i muscoli più severamente colpiti dall’evoluzione della malattia, quelli meno colpiti, il ruolo svolto dalla durata di malattia sull’evoluzione/non evoluzione del quadro ipostenico.
Le osservazioni dei dati di funzionalità motoria, raccolti tramite la scala MFM (Motor Function Measure, validata a livello internazionale) , mostrano come il valore ottenuto alla subscala D1 (ortostatismo e trasferimenti) sia in assoluto quello più suggestivo della condizione motoria di un paziente affetto da distrofia muscolare dei cingoli da deficit di calpaina3. Infatti, un valore di D1 < a 12 corrisponde a perdita del cammino in tutti i soggetti, mentre un valore compreso tra 40 e 60 di punteggio totale della scala si associa ad una importante variabilità clinico funzionale, comprendendo pazienti con cammino autonomo e pazienti costretti alla carrozzina.
Queste preliminari osservazioni potranno essere preziose per le successive osservazioni/correlazioni dei dati di imaging muscolare.
Raccolta dei dati di Imaging muscolare
In tutti i pazienti verranno effettuate acquisizioni a 3T della muscolatura del cingolo scapolare e della muscolatura prossimale degli arti superiori ed inferiori , utilizzando sequenze pesate in T1, T2, inversion recovery, 3 point Dixon come precedentemente descritto (Hollingsworth KG et al 2012, Astrea G 2009, Astrea G 2012). Il grado di interessamento muscolare verrà valutato con criteri semiquantitativi utilizzando scale precedentemente pubblicate (Mercuri E. et al, 2002). Verrà anche valutata la possibilità, in un sottogruppo di pazienti, di applicare parametri quantitativi. Lo studio verrà completato con sequenze di perfusione musoclare.
In un sottogruppo limitato di pazienti condurremo inoltre uno studio esplorativo al fine di andare a valutare la sensibilità e specificità dello studio di Risonanza Magnetica della muscolatura respiratoria (muscoli intercostale, addominali e diaframma) e la eventuale correlazione con la funzionalità respiratoria misurata tramite spirometria e pletismografia optoelettronica.
Nel corso del 2015 sono stati effettuati i primi studi RM su pazienti affetti da distrofia muscolare sia ad eziologia nota/geneticamente determinata che ad eziologia genetica non ancora definita.
Il protocollo RM ottimizzato ad hoc per lo studio includeva sequenze:
– T1 pesate per la caratterizzazione morfologica del muscolo
– T2 pesate multi-echo per la quantificazione del parametro T2 dei tessuti
– DIXON per la separazione del segnale di acqua e grasso, con quantificazione della frazione di grasso.
– Diffusion MR spinEcho a multipli valori di b, per l’analisi strutturale del tessuto muscolare, mediante quantificazione del parametro FA (fractional anisotropy) e ricostruzioni trattografiche (DTI).
Sono stati sottoposti a RM muscolare 7 soggetti sani e 22 pazienti affetti da diverse forme di distrofia muscolare (8 deficit calpaina, 3 deficit di disferlina, 4 distrofie di Becker, 1 deficit di anoctamina, 1 deficit di β-sarcoglicano, 1 deficit di collagene tipo VI, 1 distrofia facio-scapolo-omerale, 3 pazienti con distrofia dei cingoli di ndd).
Gli esami sono stati acquisiti con scanner RM 3T ed è stata sottoposta a indagine la muscolatura della coscia. Tutti i soggetti hanno tollerato l’esame e non si sono verificate complicanze durante l’esecuzione delle indagini RM.
I dati di ciascun soggetto sono stati processati per ottenere mappe strutturali, mappe del parametro T2, mappe FA e mappe di fat-fraction (FF).
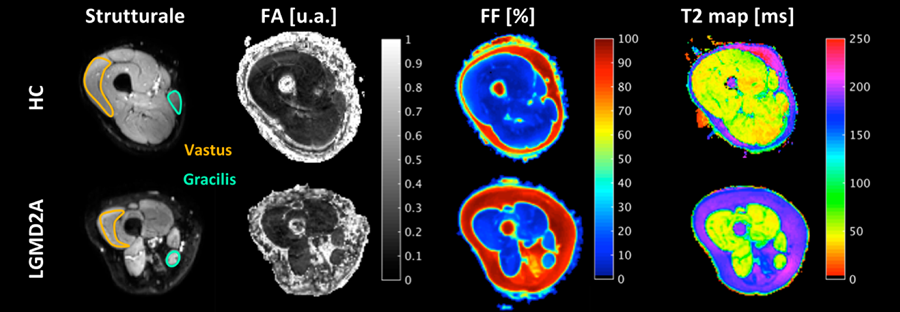
Nel campione dei sani, le misure effettuate hanno dimostrato omogeneità e ripetibilità inter-soggetto per tutte le variabili sinora considerate (FA, T2, FF).
Nel gruppo dei pazienti, tali variabili si sono dimostrate sensibili a misurare le modificazioni architetturali e strutturali del muscolo. In particolare, sono state effettuate misurazioni preliminari a livello dei muscoli vasto mediale e gracile che hanno evidenziato comportamenti e modificazioni diverse dei parametri considerati in relazione allo stato di alterazione strutturale e al gruppo muscolare considerato.
E’ stata inoltre dimostrata la fattibilità di ricostruzioni trattografiche a livello sia del tessuto muscolare sano che di quello patologico, con evidenziazione del sovvertimento architetturale delle fibre muscolari in pazienti affetti da diverse forme di distrofia.
CONCLUSIONI
The aim of this study was to apply quantitative MRI (qMRI) to assess structural modifications in thigh muscles of subjects with limb girdle muscular dystrophy (LGMD) 2A and 2B with long disease duration. Methods: Eleven LGMD2A, 9 LGMD2B patients and 11 healthy controls underwent a multiparametric 3T MRI examination of the thigh. The protocol included structural T1weighted images, DIXON sequences for fat fraction calculation, T2 values quantification and diffusion MRI. Region of interest analysis was performed on 4 different compartments (anterior compartment, posterior compartment, gracilis, sartorius). Results: Patients showed high levels of fat infiltration as measured by DIXON sequences. Sartorius and anterior compartment were more infiltrated in LGMD2B than LGMD2A patients. T2 values were mildly reduced in both disorders. Correlations between clinical scores and qMRI were found. Conclu- sions: qMRI measures may help to quantify muscular degeneration, but careful interpretation is needed when fat infiltration is massive.
 Scarica il Fulltext in Inglese
Scarica il Fulltext in Inglese